近年の遺伝子工学的技術の進展に伴い生物のシステムの解明が進みつつある。ヒトゲノム計画に代表されるゲノムプロジェクトにより生物の設計図といえるゲノムDNA配列情報が明らかにされてきた。われわれの研究室では、この膨大な設計図に何がかかれているのかを解明すること、さらに分子生物学実験における精度の高い計測技術の開発を行っている。生物を超並列ナノ分子機械の集合体としてとらえた生命論への情報工学的アプローチを行い、有用な生物の開発や病因遺伝子を発見し病気を抑制するための遺伝子工学的方法論の開発を目標とする。
一度に数万の遺伝子発現量を定量可能なDNAマイクロアレイは、疾患の診断やメカニズム解明など様々な応用が期待されているが、得られる遺伝子発現プロファイルの情報量が膨大であるため、必要な情報を抽出するには工夫が必要となる。例えば、癌組織や健常組織といった、事前に何らかの臨床情報が得られている発現プロファイルの群間比較を考えると、この問題は、臨床情報を教師信号、各遺伝子の発現量を入力信号とみなすことにより、supervised learningのアルゴリズムが適用可能となり、古典的な学習分類問題に帰着する。
しかし、DNAマイクロアレイから得られるデータには実験誤差、異種細胞の混入など様々なノイズが存在し、また、同一の疾患でも異なる遺伝子の異常により生じている場合があるため、二群分類を100%の精度で行うことは困難である。そのため、単に遺伝子発現プロファイルを二群に分類するだけでなく、疾患に関して豊富な知識を持つ専門の研究者が、容易に遺伝子発現プロファイルの特徴を理解し、異常サンプルを発見するために、発現プロファイルの分布を可視化することが重要となる。一般に遺伝子発現プロファイルは非常に高次元なデータであるため、主成分分析などを用いて、次元を二次元、もしくは三次元まで削減することによって可視化が行われている。しかし、主成分分析など、従来の可視化手法は、臨床情報を利用せずに次元削減を行うため、得られる部分空間が二群間比較に最適とは限らない。
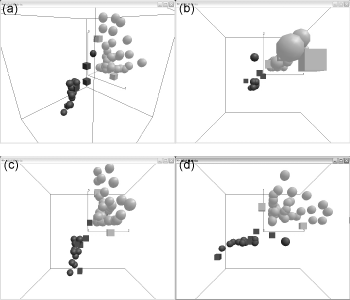
そこで、我々は、二群間比較において最も優れた成果を挙げているSupport Vector Machines (SVM)を多次元に拡張した多次元サポートベクターマシン (Multidimensional SVM, MD-SVM)を提案した。MD-SVMは主成分分析と異なり、臨床情報を利用し、二群を鮮明に分ける軸を複数抽出することにより、可視化を実現する。我々はMD-SVMを、肺ガン組織及び正常肺組織から得られた遺伝子発現プロファイルに適用し、主成分分析に比べ、二つの群が鮮明に分かれていること、また、肺ガン組織のサンプル中に存在する異常サンプルが容易に検出できることを確認した。
MD-SVMを用いて得られた肺ガンデータセットの分布.
図中の黒で示されたサンプルは正常肺組織を表し、白で示されたサンプルは肺ガン組織を表す。また、訓練データを球、テストデータを立方体で示す。なお、矢印で示されたサンプルは、他の肺ガンとは異なる、もしくは大きな実験誤差を含んだサンプルであると考えられる。